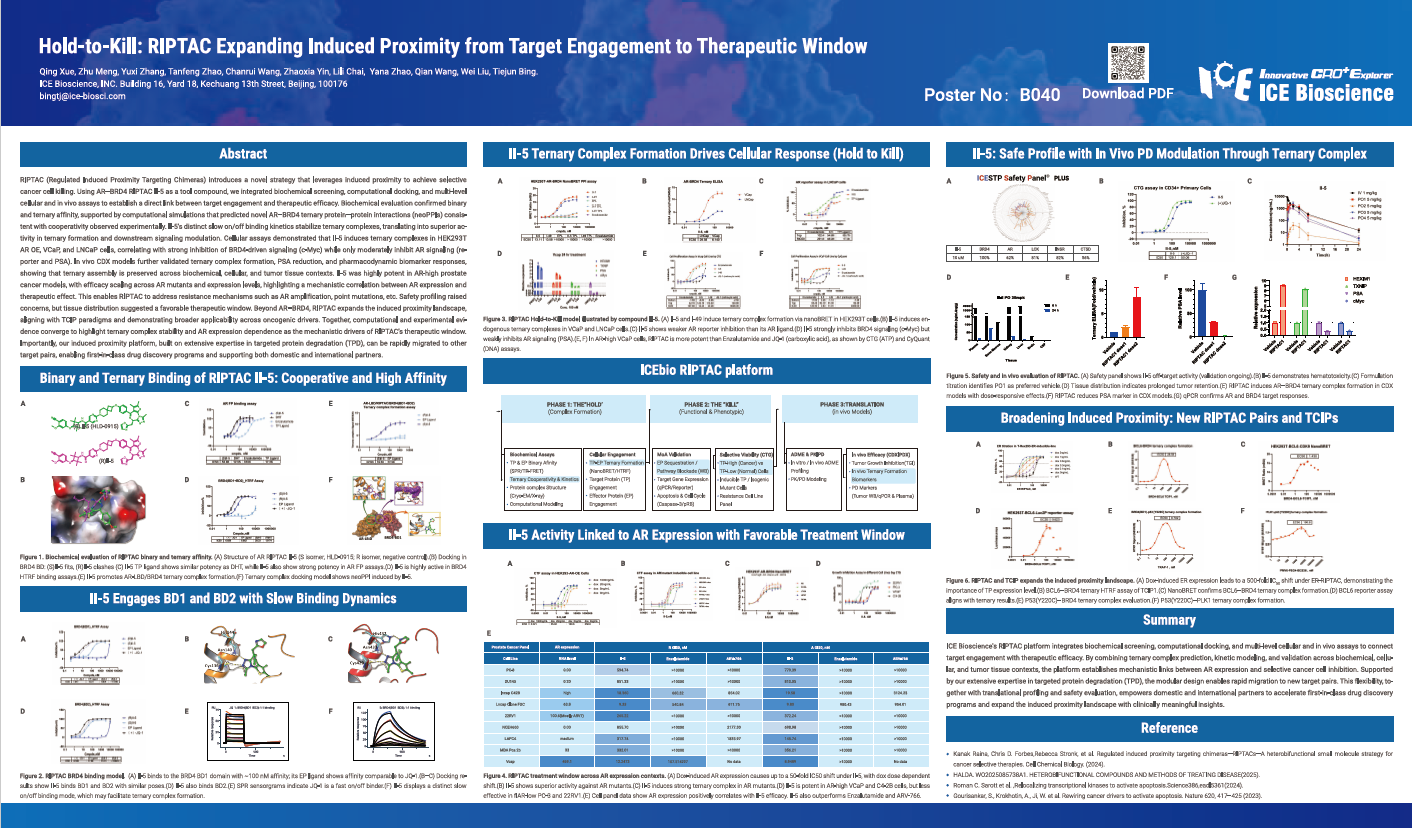
RIPTAC (Regulated Induced Proximity Targeting Chimeras) introduces a novel strategy that leverages induced proximity to achieve selective cancer cell killing. Using AR–BRD4 RIPTAC II-5 as a tool compound, we integrated biochemical screening, computational docking, and multi-level cellular and in vivo assays to establish a direct link between target engagement and therapeutic efficacy. Biochemical evaluation confirmed binary and ternary affinity, supported by computational simulations that predicted novel AR–BRD4 ternary protein–protein interactions (neoPPIs) consistent with cooperativity observed experimentally. II-5’s distinct slow on/off binding kinetics stabilize ternary complexes, translating into superior activity in ternary formation and downstream signaling modulation. Cellular assays demonstrated that II-5 induces ternary complexes in HEK293T AR OE, VCaP, and LNCaP cells, correlating with strong inhibition of BRD4-driven signaling (c-Myc) while only moderately inhibit AR signaling (reporter and PSA). In vivo CDX models further validated ternary complex formation, PSA reduction, and pharmacodynamic biomarker responses, showing that ternary assembly is preserved across biochemical, cellular, and tumor tissue contexts. II-5 was highly potent in AR-high prostate cancer models, with efficacy scaling across AR mutants and expression levels, highlighting a mechanistic correlation between AR expression and therapeutic effect. This enables RIPTAC to address resistance mechanisms such as AR amplification, point mutations, etc. Safety profiling raised concerns, but tissue distribution suggested a favorable therapeutic window. Beyond AR–BRD4, RIPTAC expands the induced proximity landscape, aligning with TCIP paradigms and demonstrating broader applicability across oncogenic drivers. Together, computational and experimental evidence converge to highlight ternary complex stability and AR expression dependence as the mechanistic drivers of RIPTAC’s therapeutic window. Importantly, our induced proximity platform, built on extensive expertise in targeted protein degradation (TPD), can be rapidly migrated to other target pairs, enabling first-in-class drug discovery programs and supporting both domestic and international partners.
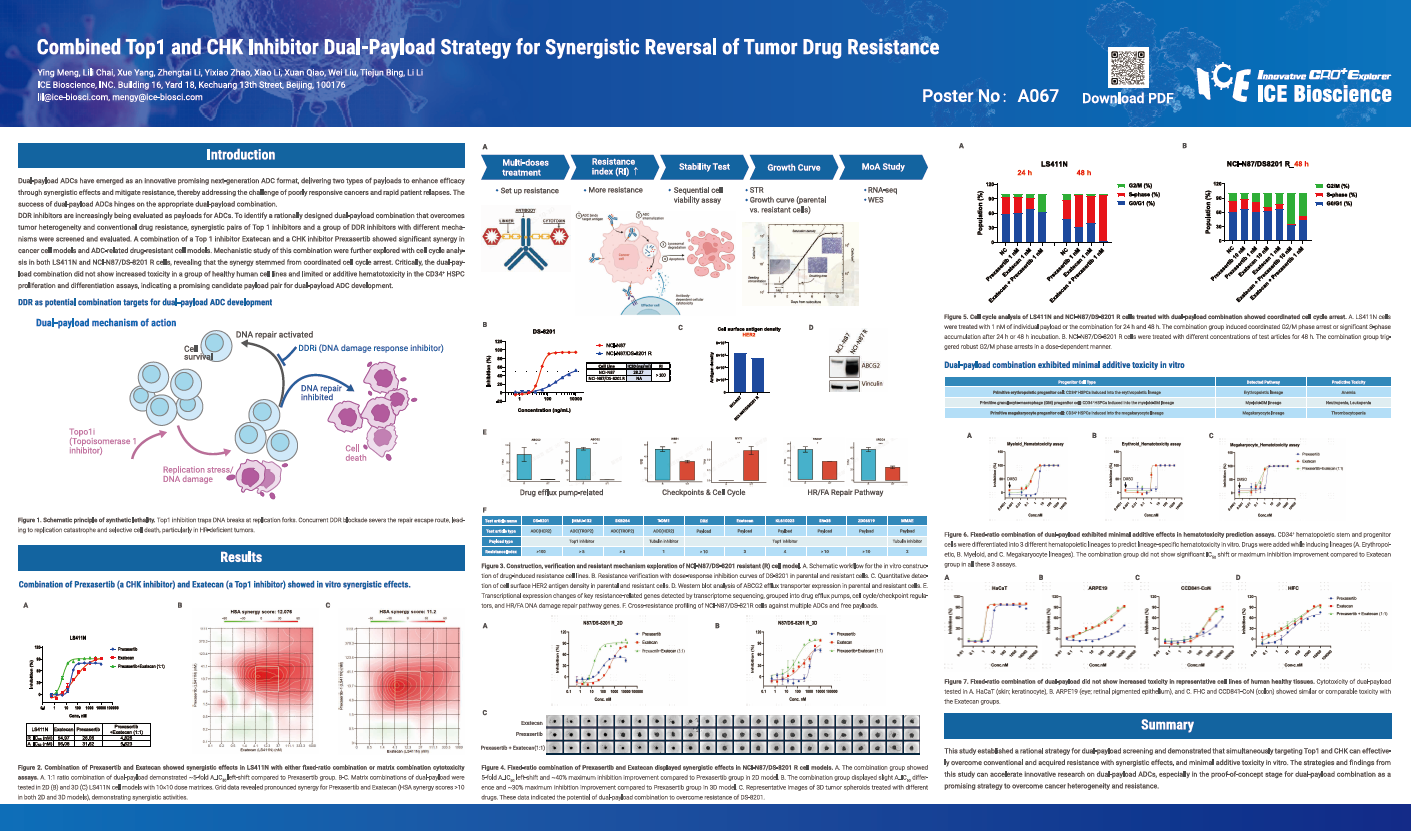
Dual-payload ADCs have emerged as an innovative promising next-generation ADC format, delivering two types of payloads to enhance efficacy through synergistic effects and mitigate resistance, thereby addressing the challenge of poorly responsive cancers and rapid patient relapses. The success of dual-payload ADCs hinges on the appropriate dual-payload combination.
DDR inhibitors are increasingly being evaluated as payloads for ADCs. To identify a rationally designed dual-payload combination that overcomes tumor heterogeneity and conventional drug resistance, synergistic pairs of Top 1 inhibitors and a group of DDR inhibitors with different mechanisms were screened and evaluated. A combination of a Top 1 inhibitor Exatecan and a CHK inhibitor Prexasertib showed significant synergy in cancer cell models and ADC-related drug-resistant cell models. Mechanistic study of this combination were further explored with cell cycle analysis in both LS411N and NCI-N87/DS-8201 R cells, revealing that the synergy stemmed from coordinated cell cycle arrest. Critically, the dual-payload combination did not show increased toxicity in a group of healthy human cell lines and limited or additive hematotoxicity in the CD34⁺ HSPC proliferation and differentiation assays, indicating a promising candidate payload pair for dual-payload ADC development.
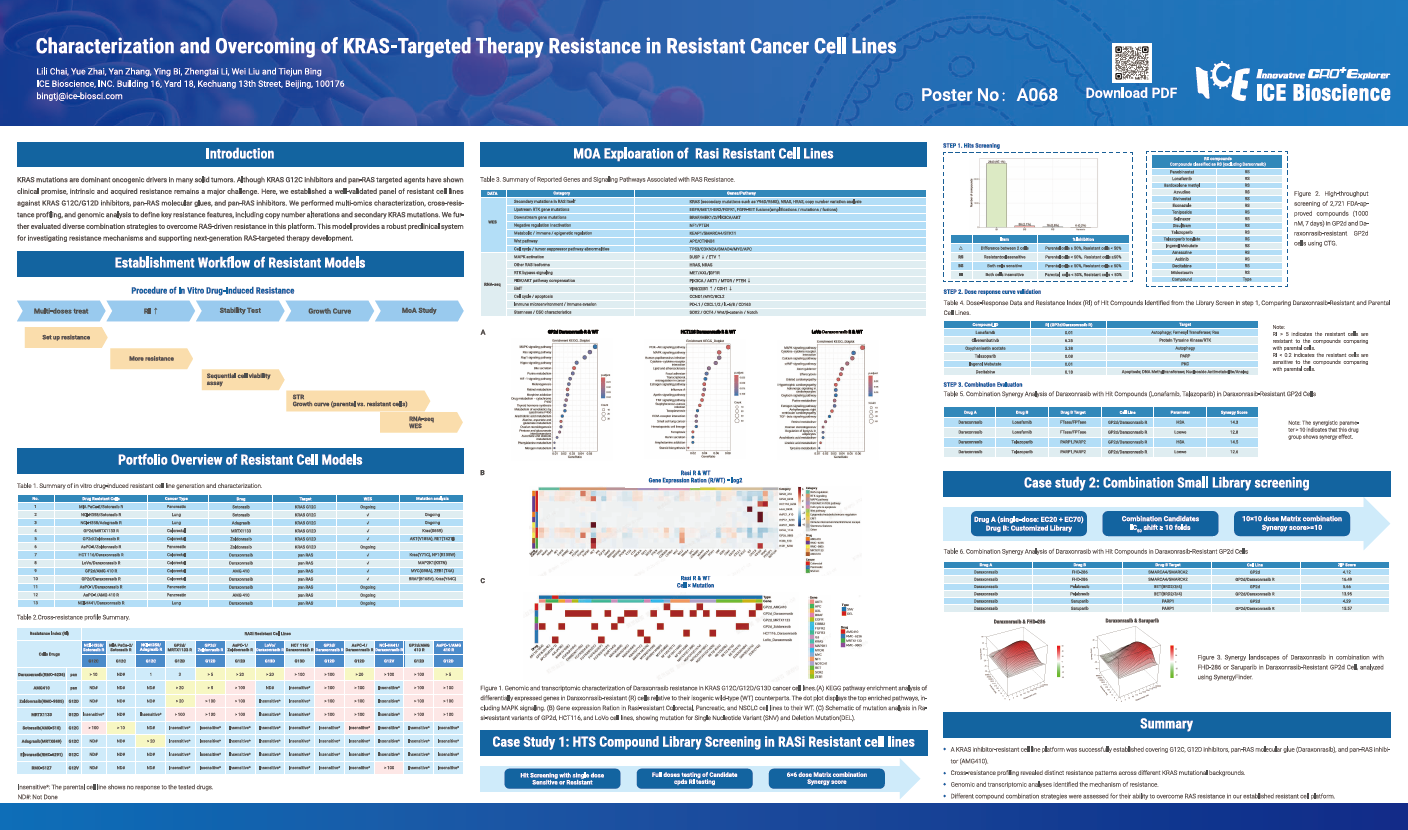
KRAS mutations are dominant oncogenic drivers in many solid tumors. Although KRAS G12C inhibitors and pan-RAS targeted agents have shown clinical promise, intrinsic and acquired resistance remains a major challenge. Here, we established a well-validated panel of resistant cell lines against KRAS G12C/G12D inhibitors, pan-RAS molecular glues, and pan-RAS inhibitors. We performed multi-omics characterization, cross-resistance profiling, and genomic analysis to define key resistance features, including copy number alterations and secondary KRAS mutations. We further evaluated diverse combination strategies to overcome RAS-driven resistance in this platform. This model provides a robust preclinical system for investigating resistance mechanisms and supporting next-generation RAS-targeted therapy development.

RIPTAC (Regulated Induced Proximity Targeting Chimeras) introduces a novel strategy that leverages induced proximity to achieve selective cancer cell killing. Using AR–BRD4 RIPTAC II-5 as a tool compound, we integrated biochemical screening, computational docking, and multi-level cellular and in vivo assays to establish a direct link between target engagement and therapeutic efficacy. Biochemical evaluation confirmed binary and ternary affinity, supported by computational simulations that predicted novel AR–BRD4 ternary protein–protein interactions (neoPPIs) consistent with cooperativity observed experimentally. II-5’s distinct slow on/off binding kinetics stabilize ternary complexes, translating into superior activity in ternary formation and downstream signaling modulation. Cellular assays demonstrated that II-5 induces ternary complexes in HEK293T AR OE, VCaP, and LNCaP cells, correlating with strong inhibition of BRD4-driven signaling (c-Myc) while only moderately inhibit AR signaling (reporter and PSA). In vivo CDX models further validated ternary complex formation, PSA reduction, and pharmacodynamic biomarker responses, showing that ternary assembly is preserved across biochemical, cellular, and tumor tissue contexts. II-5 was highly potent in AR-high prostate cancer models, with efficacy scaling across AR mutants and expression levels, highlighting a mechanistic correlation between AR expression and therapeutic effect. This enables RIPTAC to address resistance mechanisms such as AR amplification, point mutations, etc. Safety panel profiling indicates an overall favorable profile with limited off-target activity. Beyond AR–BRD4, RIPTAC expands the induced proximity landscape, aligning with TCIP paradigms and demonstrating broader applicability across oncogenic drivers. Together, computational and experimental evidence converge to highlight ternary complex stability and AR expression dependence as the mechanistic drivers of RIPTAC’s therapeutic window. Importantly, our induced proximity platform, built on extensive expertise in targeted protein degradation (TPD), can be rapidly migrated to other target pairs, enabling first-in-class drug discovery programs and supporting both domestic and international partners.
Address: Bldg 16, Yd 18, Kechuang 13th St, Etown, Tongzhou Dist, Beijing, 100176, China
Email: marketing@ice-biosci.com
Tel:+86-10-67809840


